


Departments of Dermatology, 1Henan Provincial People’s Hospital, No.7 Weiwu Road, Zhengzhou 450003, Henan, 2Zhengzhou University People’s Hospital, Zhengzhou, Henan, 3Xinhua Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, and 4Kaifeng Second People’s Hospital, Kaifeng, Henan, China. *E-mail: aypyslm@163.com; lizhenlu@sohu.com
#These authors contributed equally to this paper.
Lentigines are flat or slightly raised, small, black-brown macules with a clearly defined edge (1). Histologically, increased numbers of melanocytes and elevated amounts of melanin can be observed. In some conditions, lentigines are associated with multisystem syndromes, which usually present symptoms at birth or early childhood, such as Peutz-Jeghers syndrome, Noonan syndrome and LEOPARD syndrome (1–4).
SASH1 is a member of the SLY-family of signal adapter proteins, and serves as a candidate tumour suppressor in breast and colon cancer (5, 6). Heterozygous SASH1 mutations were initially reported to be associated with dyschromatosis universalis hereditaria (DUH), which is predominantly characterized by generalized mottled hyper- and hypo-pigmentation (7).
Recently, several cases of SASH1-related lentiginous phenotype have been reported in succession (8–10). This report describes the clinical and molecular delineation of a Chinese family with multiple lentiginous phenotypes, and the successful treatment of facial lentigines with a 755-nm Q-switching alexandrite laser.
This study was approved by the Institutional Review Board of Henan Provincial People’s Hospital, Henan, China, and was conducted in accordance with the principles of the Declaration of Helsinki. After obtaining written consent, the proband, his parents, his grandparents and 100 ethnic-matched healthy controls were indexed in this study.
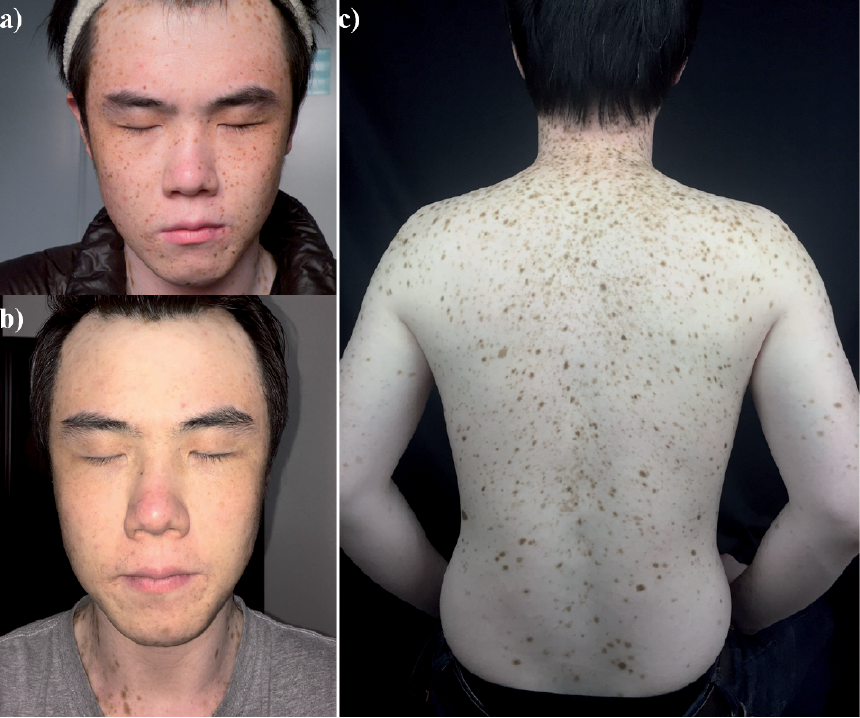
The proband is a 27-year-old man from Henan Province, China, who was referred to our department in June 2015 with multiple “freckles” on the forehead since 18 months of age. The lesions gradually increased and spread to the patient’s entire body by the age of 10 years. Dermatological examination revealed multiple lentigines on the trunk, limbs and face (Fig. 1). His mother has a similar lentiginous phenotype. No mental abnormalities, facial malformation, or cardiac defects were present in the patient or his mother.
Fig. 1. Clinical picture of the proband with SASH1 mutation. (a) Multiple lentigines on the face. (b) Facial appearance after treatment with a 755 nm Q-switching alexandrite laser. (c) Multiple lentigines on the trunk and limbs. A written permission is given by the patient to publish these photos.
Peripheral blood samples from the indexed subjects were collected for molecular analysis. Genomic DNA was extracted using TIANamp Blood DNA Kit (Tiangen Biotech, Beijing, China). Primers flanking all coding regions of SASH1 were designed using software Primer Premier 5.0 (Premier Biosoft International, Palo Alto, CA, USA). The extracted genomic DNA samples were amplified by PCR. Purified PCR products were sequenced directly using an Abi Prism®3730 automated sequencer (Applied Biosystems, Foster City, CA, USA). Mutation was described by comparison with the NCBI cDNA reference sequence: NM_015278.3.
A novel heterozygous missense mutation c.1519T>G (p.Ser507Ala) in SASH1 was identified in the proband and his affected mother. Moreover, mutation p.Ser507Ala was not identified in his unaffected father, his grandparents or in 100 normal controls (Fig. S1). This is a de novo mutation. Online in silico programmes, such as PolyPhen2 (http://genetics.bwh.harvard.edu/pph2) and SIFT (http://sift.bii.a-star.edu.sg), were applied to predict the potential impact of an amino acid substitution. Mutation p.Ser507Ala was predicted to be “damaging”, with a score of 0.002 in SIFT, and to be “probably damaging”, with a score of 0.994 in Polyphen2, respectively. Moreover, a 755-nm Q-switching alexandrite laser is effective for the treatment of facial lentigines after first treatment (Fig. 1c). The proband was followed up for 2 months, but longer-term follow-up is needed to validate the effect.
SASH1 is located on chromosome 6q24.3, and encodes SASH1 protein with a length of 1230 amino acids. SASH1 contains a SLY domain (amino acid residue 382–536), a SH3 domain (amino acid residue 539–594), and 2 SAM domains (amino acid residue 614–676 and 1155–1220, respectively) (7).
Zhou et al. (7) screened selected genes in a 10.2-Mb region on chromosome 6q24.2–q25.2 in a patient with DUH phenotype and successfully identified the causal gene SASH1 located at 6q24.3. They then investigated the pathogenic mechanism of SASH1 in regulating pathological cutaneous pigmentation and revealed that mutated SASH1 genes can enhance expression of premelanosome and mature melanosome resident proteins (TYRP1 and Pmel17), and melanosome transport protein (Rab 27a) to increase the biosynthesis and transport of melanin in melanocytes. Furthermore, SASH1 regulates melanocyte transepithelial migration through the Gαs–SASH1–IQGAP1–E-cadherin dependent path-way (7).
To date, 7 different missense SASH1 mutations have been identified as responsible for pigmentary inherited skin disorders; 3 heterozygous mutations (p.Glu509Lys, p.Leu515Pro and p.Tyr551Asp) were found to contribute to a DUH phenotype; mutation p.Ser519Asn, p.Ser513Arg and 1527_1530dupAAGT (p.Leu511Lysfs*21) were found to result in a similar phenotype to that in the present case, with multiple lentigines prominent in sun-exposed areas with or without dyschromatosis (9, 10); and, notably, a homozygous p.Glu617Lys mutation was found to be responsible for a new genodermatosis with multiple hyperpigmented macules, palmoplantar keratoderma and skin carcinoma (8).
Considering that: (i) the newly identified p.Ser507Ala substitution is located in the highly conserved SLY domain; (ii) functional consequence predictions with PolyPhen2, SIFT were deleterious and damaging; (iii) the mutation is absent from 200 alleles of 100 normal controls; and (iv) 5 similar pathogenic mutations (p.Glu509Lys, p.Ser513Arg, p.Leu515Pro, p.Ser519Asn and p.Tyr551Asp) clustered in the same domain have been reported to cause pigmentary abnormalities (including lentigines) (9), it is thought that the novel mutation observed by us was probably the pathogenic mutation responsible for this patient developing typical clinical features of multiple lentigines,
In conclusion, this study investigated a pedigree with lentiginous phenotypes and successfully identified novel pathogenic mutation in a hotspot mutation region of SASH1 (SLY domain), which further verifies that SASH1 is the causal gene responsible for lentiginous phenotypes with or without dyschromatosis.
The authors would like to thank all subjects for their ongoing participation in this study. This study was funded by a grant from the National Nature Science Foundation of China (81472867).
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize