


1Department of Dermatology, St George Hospital, 2Faculty of Medicine, University of New South Wales, Sydney, 3Department of Paediatrics, Murdoch Children’s Research Institute/University of Melbourne, Royal Children’s Hospital, Parkville, and 4Faculty of Medicine, Monash University, Eastern Health, Box Hill, Australia
Epidermolysis bullosa simplex (EBS) is a rare heritable skin fragility disorder, most commonly caused by dominant mutations in KRT5 and KRT14. EBS shows clinical heterogeneity with localised, intermediate and generalised severe forms, which tend to correlate with the location and nature of the disease causing mutations. We therefore aimed to identify the KRT5 and KRT14 mutations in patients diagnosed with EBS in Australia, and explore in depth the genotype to the phenotype correlations in patients with novel variants. Australian patients who were diagnosed with EBS after referral to the Australian National Diagnostic Laboratory for EB were offered mutation screening in the KRT5 and KRT14 genes. From this, 32 different mutations in KRT5 and KRT14 were identified within 39 of 52 pedigrees. Ten of these mutations from 9 different pedigrees were novel, a further fatal case caused by KRT5 E477K is reported and in addition the third reported case of digenic inheritance in EBS was also observed.
Key words: epidermolysis bullosa simplex; keratin 5; keratin 14.
Accepted May 30, 2017; Epub ahead of print May 31, 2017
Acta Derm Venereol 2017; 97: XX–XX.
Corr: Professor Dedee F. Murrell, Department of Dermatology, Ground floor James Law House, St George Hospital, Gray Street, Sydney, New South Wales, Australia. E-mail: d.murrell@unsw.edu.au
Epidermolysis bullosa simplex (EBS) is a heritable disease most commonly caused by dominant-negative mutations in the genes encoding keratin 5 and keratin 14 (KRT5 and KRT14) (1). Keratin 5 and 14 dimerise to form intermediate filaments, which provide structure, strength and flexibility to the keratinocyte cytoskeleton. When compromised, they are susceptible to mechanical stress, leading to the fracture of basal keratinocytes and subsequent blistering of the epithelium (2).
EBS exhibits clinical heterogeneity with 3 main subtypes (3). Epidermolysis bullosa simplex-localised (EBS-loc), resembling the previously used eponymous EBS-Weber Cockayne, is the most common form and is characterised by blistering confined to soles and palms, worse with friction, trauma, heat and sweating (3). EBS-gen intermediate, resembling the eponymous EBS-Koebner, presents with more widespread blistering, but in a form that is milder than EBS-gen severe. EBS-gen severe, resembling the eponymous EBS-Dowling Meara, involves widespread clustered blistering, erosions, scarring, milia and can involve the mucous membranes, hair and the nails (4).
The different EB subtypes tend to correlate with the location and nature of the inherited mutation (5). The highly conserved ends of the rod domain (helix boundary motifs) of KRT5 and KRT14 are crucial for correct keratin filament assembly and mutations in these regions often result in severe blistering, whereas mutations outside of these regions tend to cause milder phenotypes of EBS (6). Despite this knowledge, genotype–phenotype predictions must be performed with caution, as the process of gene expression is complex and can be influenced by other genetic and epigenetic factors (7). It is therefore essential that genotype–phenotype correlations are published, especially those of patients with novel mutations and unexpected phenotypes, as this can lead to a better understanding of the pathogenesis of EBS, clarify prognosis and assist in prenatal testing (8).
We aim to review the KRT5 and KRT14 mutations known to cause EBS in the Australian population and look in depth at the pedigrees in which we found novel mutations.
We performed a cross-sectional analysis using patient data from both the Australian National Diagnostic Laboratory Database for EB (9) and the Australasian EB Registry (10). Patients included in the study were located in Australia and had a confirmed diagnosis of EBS based on a combination of clinical history, immunofluorescence mapping (IFM) and electron microscopy (EM). Data gathered included sequencing results of both KRT5 and KRT14 and clinical phenotypes. If phenotypic data was unclear, patient records were reviewed and the patient was contacted and reviewed in person if further information was needed.
Mutation screening was performed on blood samples obtained with informed consent. Genomic DNA was extracted using a commercial kit (Qiagen Blood DNA Kit; Qiagen, Hilden, Germany) and analysed by bi-directional sequencing of exons 1–9 of KRT5 and exons 1–8 of KRT14. The study was approved by the South Eastern Sydney Local Health District Human Research Ethics Committee.
The Australian National Diagnostic Laboratory Database for EB and the Australasian EB Registry contained a total of 408 patients with confirmed EB. One hundred and sixty-five patients had EBS, of which probands and relatives from 52 unrelated pedigrees had sequencing of KRT5 and KRT14 performed.
Mutations in KRT5 and KRT14 were identified in 39 of the 52 pedigrees, equating to 75% of patients. Thirty-two different mutations were found, 17 in KRT5 (53%) and 15 in KRT14 (47%). One patient had digenic inheritance of one mutation in each of KRT5 and KRT14 causing EBS, the third reported digenic case of EBS published thus far (11). Mutations in KRT5 and KRT14 found in our study as well as all published mutations in KRT5 and KRT14 so far documented can be seen in Fig. 1 (data collected from http: //www.interfil.org (12)). The mutational data of the 52 pedigrees including the recurrent and novel mutations can be seen in Table SI. The cases with novel mutations are discussed below.
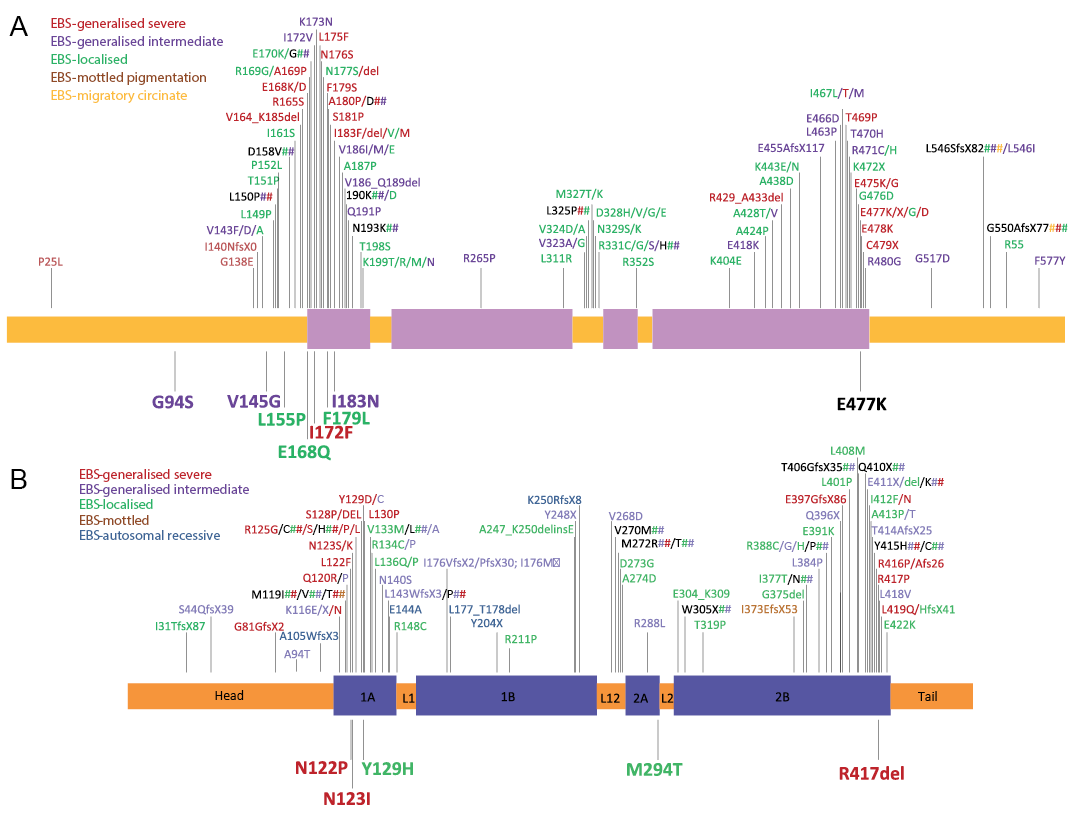
Fig. 1. Schematic diagram showing the distribution of identified amino acid variations caused by mutations within the KRT5 (a) and KRT14 (b) gene and location in the polypeptide. The rod domain of the keratin polypeptide consists of 4 alpha-helical domains (1A, 1B, 2A and 2B) separated by non-helical linker domains (L1, L12 and L2). Novel mutations described in our study are marked below the polypeptide while all known EBS causing mutations to date are marked above the peptide. The phenotypes reported to be associated with the mutations are indicated by the different colours. The further lethal case of E477K has been underneath added in black (data collected from http: //www.interfil.org (12)).
Twenty-one pedigrees were affected with EBS-loc, 10 with EBS-gen intermediate, and 21 EBS-gen severe. The clinical phenotypes of the 9 pedigrees with novel mutations are summarised in Table I.
Table I. The phenotypes of Australian patients affected by epidermolysis bullosa simplex caused by novel mutations in keratin 5 and 14
We present 52 Australian pedigrees affected by EBS sequenced for KRT5 and KRT14 mutations. Mutations in KRT5 and KRT14 were detected in 75% of the pedigrees; similar to rates in other studies (1, 8). The remaining cases are possibly due to mutations in intronic or promotor regions, or other genes implicated in EBS such as PLEC1, encoding plectin (8). Fifty-three percent of the mutations were found in KRT5 and 47% in KRT14, confirming the trend of approximately equal contribution of both genes in the disease as seen in other studies (8, 13).
As demonstrated in Fig. 1, mutations causing EBS-loc occur sporadically across the keratin 5 and 14 polypeptides. Mutations associated with the EBS-gen severe were most commonly clustered at the helix boundary motifs, the helix initiation (HIP) and termination peptides (HTP) regions, which are critical for normal keratin filament formation (14). In most other cases, phenotypes correlated with the location of the mutations and were in agreement with previous reports.
In one of our pedigrees, EB361, digenic mutations were observed where one mutation in KRT5 and another in KRT14 worsened the consequences of each separate mutation, resulting in disease (11). We present another pedigree EB534 where genetic variants may have had a disease-modifying role (Fig. S1). The proband and his father had the novel KRT5 mutation F179L located in the highly conserved 1A domain. The father was unaffected, while the proband suffered from localised blistering on the hands and feet that developed in childhood. Interestingly, the proband had several maternally inherited single nucleotide polymorphisms (SNPs) in KRT5, which were absent in the father. It is known that SNPs can affect gene function and phenotype (15, 16). Therefore, this case potentially suggests that maternally inherited SNPs found in the proband could influence the expression of F179L, producing disease. However, confirmation of this would require reproduction by in vitro experiments.
As well as EB534, two other pedigrees with novel mutations correlated with the EBS-loc phenotype. The proband in EB516, a Caucasian female, presented at age 21 with occasional blistering of the soles and palms since the age of 12 months. Mutation analysis revealed two variants in KRT14; p.Y129H and p.V452I. V452I is unlikely to be pathogenic due to its location in the tail region, coupled with an in silico analysis (SIFT, PolyPhen2) predicting that it is not likely to affect protein function. Y129H is in the highly conserved 1A region, and is likely to be pathogenic as two other mutations have been reported at this location: p.Y129C and p.Y129D, resulting in EBS-gen intermediate and EBS-gen severe, respectively (5, 17). In this case, the mutation results in an amino acid chemically similar to the original, which may explain the mild phenotype.
The proband of EB330 was a Caucasian female who suffered from mild blistering confined to the palms and soles since infancy and was diagnosed with the EBS-loc phenotype. Genetic screening of the proband and her similarly affected daughter and father revealed the heterozygous mutation p.E168Q in KRT5. This change of the charged glutamic acid (E) to the polar uncharged glutamine (Q) is located in the conserved HIP region of KRT5, where two other mutations previously described have resulted in EBS-gen intermediate and EBS-gen severe (13, 18). The reason for the relatively mild phenotype despite the change in polarity caused by the amino acid substitution in our patient is unclear, and suggests an influence of other genetic and epigenetic factors.
Two novel mutations found in the pedigrees EB74 and EB294 resulted in the EBS-gen intermediate phenotype. In EB74, the proband presented with blisters as a neonate at acral sites and in the mouth (Fig. S2). At 16 years of age, she had blisters on the fingers and toes, as well as erosions in the mouth, milia and dystrophic nails. Genetic screening of the proband and the parents revealed the de novo heterozygous mutation p.I183N in the conserved 1A region of KRT5. The EBS-gen intermediate phenotype could be explained by a significant difference in polarity and hydrophobicity of the substituted amino acid, isoleucine. This location appears to be sensitive to sequence variations as several other mutations have been described at this site (14, 19, 20).
EB294 is a large pedigree (Fig. S2), with many affected family members having a similar history of generalised blistering at birth, improving with age to involve only frequently traumatised sites such as the feet, hands and under the bra. All affected members with genetic screening performed had two in cis novel mutations in KRT5 exon 1; G94S (polar uncharged glycine to polar uncharged serine) and V145G (nonpolar hydrophobic valine to polar uncharged glycine) in the head domain (see Fig. 1a). Both mutations were predicted to be damaging by PolyPhen2 analysis, although the amino acid properties are more different in V145G, which is in the same region as other disease-causing variants. In this pedigree, the involvement of two mutations rather than one may explain the generalised phenotype, however as the mutations were always inherited in cis we cannot be certain which of the two mutations, or both, causes disease.
Four pedigrees with novel KRT5 or KRT14 mutations were associated with the EBS-gen severe phenotype. As a teenager, the proband of the pedigree EB70 suffered from severe generalised blistering, most affecting the groin, lower abdomen and genitals. This improved with age to mostly only affect the palms and soles. Mutation screening revealed the KRT5 mutation p.I172F, changing the non-polar hydrophobic isoleucine to non-polar hydrophobic phenylalanine, located in the conserved helix initiation peptide (HIP) region. This mutation was passed down to his daughter, who also developed EBS-gen severe (Fig. 2). A substitution at the same nucleotide position p.I172V, in which valine is also hydrophobic and non-polar, has been described in patients with localised EBS (8). The severe phenotype in this case is likely caused by the substantially different structure of the phenol ring in phenylalanine, the amino acid substituted. The deleterious effect in protein function was confirmed by in silico analysis (PolyPhen2 score 0.999, SIFT score 0.03).
Fig. 2. Clinical photos of daughter of EB70. (a) Herpetiform truncal plaques. (b) Blisters and hyperkeratosis of feet. (c) Blisters around warts on fingers.
EB503 is the fourth child of non-consanguineous Caucasian parents, who at delivery, demonstrated extensive blistering of her hands and feet (Fig. 3). Further lesions developed progressively post-partum at sites of friction, including the buttocks, groin, umbilicus, axillae and neck, while mucosal surfaces and hair were spared. At the age of 5, she continues to develop blisters at sites of minimal trauma, although less than before (see Fig. 3). She has also developed palmoplantar hyperkeratosis and onychogryphosis. Mutational screening of the proband and parents revealed a novel de novo mutation in KRT14, c.1250_1252delGCC and a second paternally inherited variant in KRT14, p.A413T. The 2B region of the KRT14 protein, within which our patient’s de novo mutation was found, is one of the 4 alpha helices that comprise the coiled-coil rod domain. The deletion of 3 base pairs GCC in CGC CTG coding for arginine and lysine to CTG results in the removal of a single amino acid (arginine) which, although maintaining the reading frame, would disrupt the helical structure. Few cases of EBS have previously been described with in-frame base pair deletions in KRT14 as found in our case (1, 13, 21–23). The pathogenic contribution of the KRT14 variant, 1237G>A (A413T) in our case remains unclear, but given that the unaffected father carried the A413T variant, it may be a recessive trait or co-dominant mutation. Clinically this variant, more common in Asian populations, has been reported to be associated with a range of presentations (24).
Fig. 3. Clinical photos of EB503 with de novo mutation KRT5 R417del in 2B region and KRT5 A413T. (a) Early degloving changes to feet. (b) Herpetiform blisters on knee. (c–e) Progression of hand blisters to a keratoderma with age.
Two other pedigrees affected by EBS-gen severe, EB307 and EB394, harboured the de novo mutations p.N123I and p.L122P, respectively, in the highly conserved HIP region of KRT14 (Fig. 1b). Other missense mutations at the same nucleotide positions with different amino acids have been described in patients also with EBS-gen severe (1, 5, 8, 13, 19, 25–28). This confirms that this region is highly conserved and therefore any substitution here is likely to cause disease.
In 8 pedigrees, mutations were identified involving the highly conserved arginine at p.125 (p.Arg125) within the HIP of KRT14. In these cases arginine was substituted with either a cysteine or histidine residue. These were all associated with EBS-gen severe, and accounted for 40% of EBS-gen severe cases our cohort, which is consistent with the current literature (www.interfil.org (12)). The relatively high frequency of this mutation is due to the hypermutable CpG dinucleotide, which is conserved in all type 1 keratins (8), and the resulting severe phenotype of EBS is due to the important role of this specific arginine residue in keratin intermediate filament assembly and function (29).
In our patients with EBS-gen severe, two were found to harbour the p.E477K de novo mutation in KRT5, which has recently been associated with high rates of lethality (30). Sathishkumar et al. (30) found that 5 out of 37 cases with EBS-gen severe died within 6 months of birth, all of whom had the mutation p.E477K in KRT5. Our patient EB266 died 36 days after birth, while EB256 survived despite severe generalised blistering including non-healing lesions, hyperkeratosis and thick nails on hands and feet. When seen at the age of 3, the proband’s weight and height were considerably below the 1st percentile with some developmental issues, especially in gross motor and balance. This supports the claim that p.E477K in KRT5 is strongly associated with significant morbidity and mortality due to the change of the second glutamate residue of the most evolutionarily conserved KLLEGE motif as the acidic glutamate is substituted to basic lysine (31).
In the majority of our patients we noticed improvement of symptoms with age. Further research is required to determine if there are other disease-modifying genes, or protein interactions that affect phenotype with age.
In summary, this study adds a further 10 novel mutations to the catalogued genotype-phenotype correlations in EBS and demonstrates a potential modifying effect of SNPs on the phenotype. We therefore support the notion of full DNA sequencing of both KRT5 and KRT14 genes so as to not miss any variants in the genes contributing to the phenotype. Patients with no KRT5/14 mutations found must then be screened for mutations in other genes such as PLEC in order to understand the pathogenesis of EBS further. This, we hope, will improve our understanding of the disease and aid genetic counselling. Subject to further research, our data can hopefully also prove useful for future personalised therapeutic strategies (32).
We would like to thank Valentine Hyland, Molecular Genetics Laboratory, Royal Brisbane Hospitals Campus, for assisting us in genetic screening of KRT5 and KRT14 genes in EBS patients.
This study was supported by the Australasian Blistering Diseases Foundation www.blisters.org.au. The Independent Learning Program of UNSW Australia supported ENK to work with DFM.
The authors declare no conflicts of interest.


 Click to show fullsize
Click to show fullsize Click to show fullsize
Click to show fullsize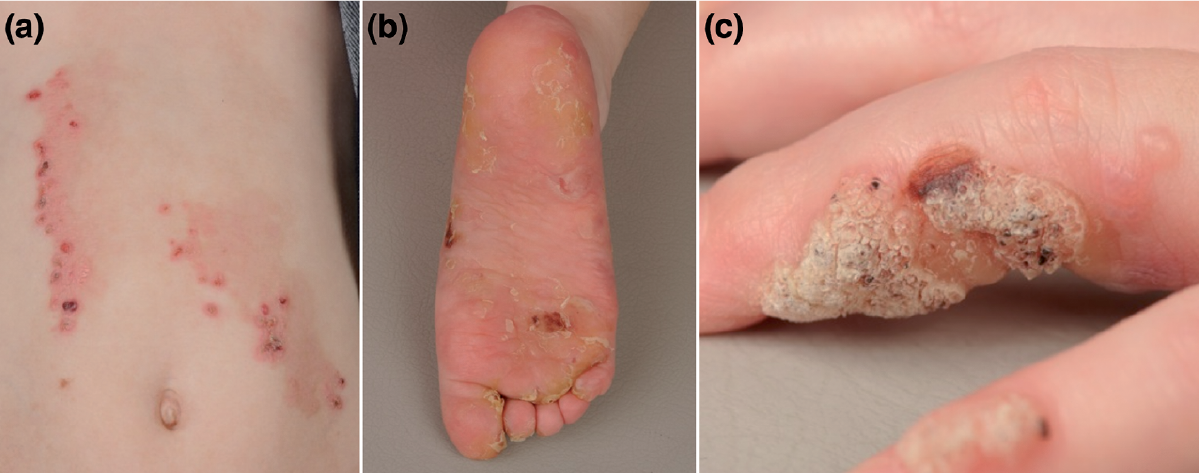 Click to show fullsize
Click to show fullsize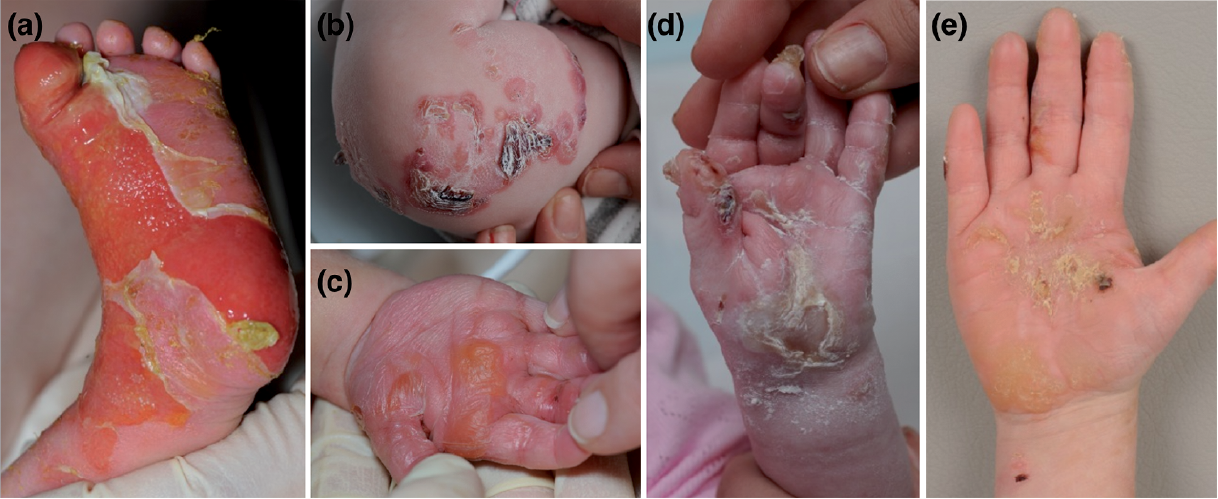 Click to show fullsize
Click to show fullsize