


1Laboratory of Molecular and Cell Biology and 31st Dermatology Division, Istituto Dermopatico dell’Immacolata-IRCCS, and 2Genetic and Rare Diseases Research Area, Bambino Gesù Children’s Hospital-IRCCS, Rome, Italy
#These authors share last authorship.
Circulating anti-type VII collagen autoantibodies are frequently detected in patients with recessive dystrophic epidermolysis bullosa (RDEB). However, evidence supporting their pathogenic role in inducing epidermolysis bullosa acquisita (EBA) has been provided for only one individual with dominant dystrophic epidermolysis bullosa (DDEB). We describe here a patient who presented with dystrophic toenails since early childhood and developed trauma-induced skin blisters and oral erosions at age 26 years. Direct immunofluorescence showed IgG deposits with a u-serrated pattern along the cutaneous basement membrane zone, while no change in the expression of collagen VII could be detected by antigen mapping. High-titre anti-collagen VII antibodies were detected by enzyme-linked immunoassay (ELISA). In parallel, sequencing of epidermolysis bullosa (EB) genes identified compound heterozygous COL7A1 missense c.410G>A (p.Arg137Gln) and splicing c.3674C>T (p.Ala1225_Gln1241del) mutations, previously unrecognized in dystrophic epidermolysis bullosa (DEB). Thus, our patient had RDEB “nails-only” and developed mechanobullous EBA in adulthood. These data support a patho-genic role of circulating autoantibodies to collagen VII in inducing EBA in selected patients with DEB. Unforeseen worsening of skin symptoms in DEB should prompt laboratory investigations for EBA.
Key words: dystrophic epidermolysis bullosa; type VII collagen; autoantibodies.
Accepted Nov 24, 2017; Epub ahead of print Nov 28, 2017
Acta Derm Venereol 2018; 98: XX–XX
Corr: Daniele Castiglia, Laboratory of Molecular and Cell Biology, Istituto Dermopatico dell’Immacolata (IDI)-IRCCS, via dei Monti di Creta 104, IT-00167 Rome, Italy. E-mail: d.castiglia@idi.it
Type VII collagen (C7) is the major constituent of anchoring fibrils, microstructures that connect the basal lamina to the underlying mesenchymal tissue. It is formed from 3 identical alpha-chains, each consisting of a central collagenous triple helix flanked by non-collagenous domains (NC), named NC1 at the N-terminus and NC2 at the C-terminus. The large NC1 domain is subdivided into modules with similarity to adhesive proteins: 2 motifs with homology to von Willebrand factor type A domain (vWFA1 and vWFA2) and 9 to fibronectin type III (1, 2). In addition, a NC1 cysteine-rich region next to the collagen triple helix domain is recognized (1, 2). NC1 subdomains interact with many extracellular matrix proteins, including collagens I and IV, and laminin-332 (2, 3).
The crucial role of C7 in mediating dermal-epidermal adhesion is underlined by 2 subepidermal skin blistering diseases with partially overlapping clinical features, but different aetiology: dystrophic epidermolysis bullosa (DEB) and epidermolysis bullosa acquisita (EBA) (1, 4). The former is caused by dominant or recessive mutations in the gene, COL7A1, that encodes C7, while the latter results from an autoimmune reactivity mediated by circulating and tissue-bound immunoglobulin G (IgG) antibodies to C7, mainly recognizing the NC1 domain (4). In most cases DEB manifests at birth, while EBA onset usually occurs in adulthood (1, 4). These features can help in distinguishing the 2 diseases, nevertheless a large fraction of patients with recessive DEB (RDEB) develop circulating anti-C7 antibodies, as detected by enzyme-linked immunoassay (ELISA) and/or immunoblotting assays (5–7). However, these patients lack the major immunodiagnostic criterion for EBA, i.e. a linear deposition of IgG along the dermal–epidermal junction in a u-serrated pattern, as detected by direct immunofluorescence (4, 8). We report here the first case of EBA developing in a individual with RDEB with previously unrecognized pathogenic mutations affecting NC1 subdomains.
Following ethical approval and informed consent, skin biopsies from the patient and blood samples from the patient, her parents and child were obtained for standard histological and electron microscopy examinations, direct immunofluorescence (DIF), immunofluorescence (IF) antigen mapping, indirect IF (IIF) on salt-split skin, ELISA assays, immunoblotting, keratinocyte cultures, and for genetic analysis. The study was conducted in compliance with the principles of the Declaration of Helsinki.
ELISAs for detection of anti-BP230, -BP180 and -C7 circulating autoantibodies were performed using commercial kits (MBL, Naka-Ku Nagoya, Japan).
Laminin-332 was affinity-purified from the culture medium of squamous carcinoma cells (SCC) 15 (CCL 15; American Type Culture Collection, ATCC) as reported (9), and C7 was prepared from the culture medium of a human amnion epithelial cell line (CCL 25; ATCC), as described previously (10). Sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis was performed using a 6% polyacrylamide gel loaded with purified laminin-332 and C7 under reducing conditions. After transfer to polyvinylidene difluoride membrane (Immobilon-P; Millipore-Merck, Darmstadt, Germany) immunoreactivity was detected, by incubation with a 1:100 dilution of patient and control sera and, for laminin-332, monoclonal antibodies K140 (anti-β3 chain) and BM165 (anti-α3), and U46 (anti-γ2 polyclonal antibody produced in our laboratory), followed by incubation with alkaline phosphatase-labelled secondary antibodies.
Screening for mutations in EB disease genes was performed using a next-generation gene panel (Trusight, Illumina) and NextSeq™ 500 apparatus, followed by Sanger sequencing for variant validation. Mutations were numbered according to the translation initiation codon of the GenBank reference sequence NM_000094.3 for COL7A1 cDNA.
Possible consequences of the c.410G>A (exon 3) and c.3674C>T (exon 27) variants on COL7A1 pre-mRNA splicing were evaluated by reverse transcriptase (RT)-PCR analysis of the mRNA purified from patient cultured keratinocytes. cDNA was obtained with Superscript™ III RT (Invitrogen-Life Technologies) and amplified using the following primers: (F) 5’-TTGTGTTCTTACTGGATGGC (exon 2) and (R) 5’-AGAAGAAGTCACTGGTGGGC (exon 5), and (F) 5’-CCCTTGAGAGGTGACATATTC (exon 26) and (R) 5’- CCAACTTGTCCTCTCAGGCC (exon 30). PCR products were identified by sequencing analysis.
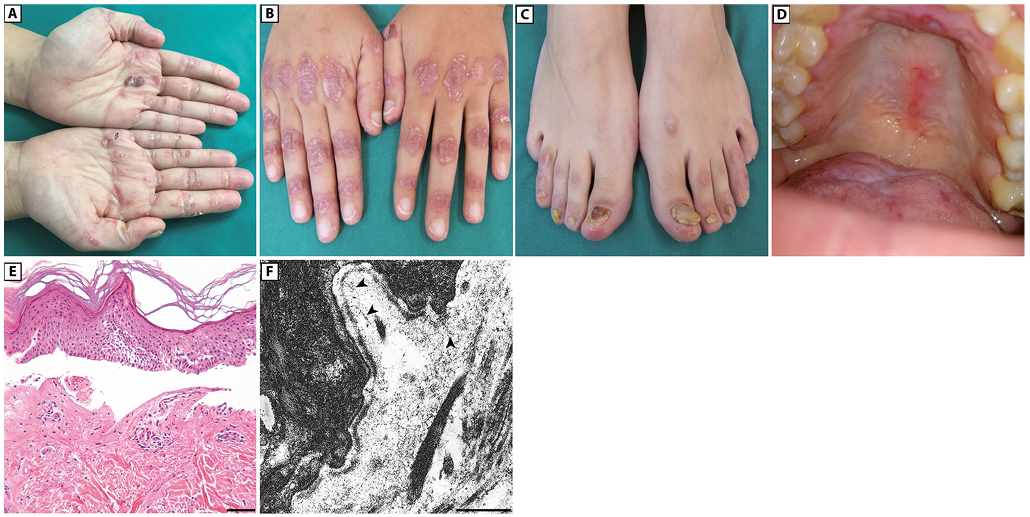
A 33-year-old woman born to non-consanguineous parents presented with a 7-year history of bullous skin eruptions mainly affecting trauma-prone areas, such as the hands, feet, elbows and knees. Physical examination revealed a few blisters, erosions and haemorrhagic crusts on her palms and soles, admixed with focal hyperkeratosis, atrophic and erythematous scars with milia on her knuckles (Fig. 1A and B) and, to a lesser extent, feet, elbows and knees, severe toenail dystrophies (Fig. 1C), and painful erosions on her oral mucosa (Fig. 1D). Her parents and her only child did not present any skin disease. The patient also reported that she had had progressive nail dystrophy limited to the big toes since early childhood; however, this condition was diagnosed as a mycosis in another hospital and not investigated further. Although history and clinical findings suggested a mechanobullous EBA, a mild form of DEB could not be excluded. Histopathological examination of a bullous lesion showed subepidermal detachment and a scant lympho-mononuclear dermal infiltrate (Fig. 1E). Ultrastructural examination of a perilesional skin biopsy showed normally structured hemidesmosomes, whilst anchoring fibrils appeared markedly hypoplastic and reduced in number (Fig. 1F). DIF examination of peri-lesional skin showed a linear deposition of IgG with a u-serrated pattern along the cutaneous basement membrane zone (BMZ) (Fig. 2A), while IF antigen mapping did not reveal changes in the expression of type VII collagen and other BMZ components (Fig. 2B). Subsequent IIF on salt-split skin demonstrated binding of IgG to the dermal side of the split skin (Fig. 2C). High levels (42 U/ml; normal value [n.v.] < 6.14) of anti-C7 autoantibodies were detected in the patient serum by a commercial ELISA. Finally, the C7 positivity was also confirmed by immunoblot (data not shown). A diagnosis of mechanobullous EBA was therefore established. Evaluation of serum reactivity to additional components of the BMZ by ELISA assays revealed positivity for both BP180 and BP230 (27.3 U/ml and 18.9 U/ml, respectively, n.v. <9). In addition, circulating IgG antibodies recognizing the α3 and β3 laminin chains were detected by immuno-blot analysis using purified laminin-332 (Fig. 2D). In parallel, screening for mutations in EB genes led to the identification of compound heterozygous variants in COL7A1: c.410G>A, p.Arg137Gln and c.3674C>T, p.Ala1225Val, inherited from the father and mother, respectively (Fig. 3A). The former variation is annotated in the ExAc database (http://exac.broadinstitute.org) with an allele frequency of 0.000131783 and without homozygous occurrence. The latter was not found in any database and is not described in the literature. Both mutations were further screened in a cohort of 62 Italian healthy individuals and never found. PolyPhen and FATHMM (Functional Analysis through Hidden Markov Models: http://fathmm.biocompute.org.uk/) predictors of mutation pathogeni-city classify the p.Arg137Gln as probably damaging and damaging, respectively, and the p.Ala1225Val as tolerated. Mutation p.Arg137Gln is within the vWFA1 (amino acids 37–201) and sequence alignment of vWFA motifs from human von Willebrand factor (vWF) and from orthologous and other paralogous proteins attests to the strict conservation of Arg137 and, thus, to its functional relevance (Fig. 3B). RT-PCR of mRNA from patient keratinocytes was then performed to investigate possible mutation effects on pre-mRNA splicing. These were not observed for the c.410G>A. Instead, a shorter cDNA product across the c.3674C>T was detected, in addition to the expected normal cDNA (Fig. 3C, left panel). Sequencing of the shorter cDNA revealed a 51-bp in-frame deletion generated by the usage of an exonic donor site formed at the mutation site (AGgcagtc→AGgtagtc) (Fig. 3C, right panel). This transcript is predicted to translate a polypeptide lacking amino acids 1225–1241 at the transition of the vWFA2 subdomain (amino acids 1054–1227) to the cysteine-rich motifs of NC1. Sequencing of the normal cDNA fragment also revealed the expression of full-length mRNA carrying the c.3674C>T, indicating that the mutation effect on splicing was leaky (not shown). Due to mutation consequences on NC1 protein sequence and structure, we concluded that our patient had the “nails-only” subtype of RDEB, as a child, and developed mechanobullous EBA in adulthood.
Fig. 1. Clinical features of the patient, and histopathology and transmission electron microscopy of patient skin. (A) Erosions and crusts on the volar aspect of patient hands. A sero-haemorrhagic blister is visible on the left palm. (B) Atrophic erythematous scars with numerous milia on hand dorsa. (C) Dystrophic toenails. (D) Multiple erosions on the palatal, gingival and tongue mucosa. (E) Histopathology shows subepidermal detachment (haematoxylin-eosin staining, bar: 50 µm). (F) Ultrastructural examination reveals markedly reduced and hypoplastic anchoring fibrils (arrowheads), bar: 0.5 µm.
Fig. 2. Direct immunofluorescence and type VII collagen expression in perilesional patient skin, indirect immunofluorescence and immunoblotting analysis with patient serum. (A) Direct immunofluorescence shows linear binding of IgG in a u-serrated pattern (arrowheads) along the cutaneous basement membrane zone (BMZ). (B) Immunofluorescence antigen mapping using LH7.2 monoclonal antibody reveals normal expression of type VII collagen along the cutaneous basement membrane zone in perilesional skin (Pt: patient; Cntr: control). (C) Indirect immunofluorescence on human salt-split skin detects IgG binding to the floor of the split (arrowheads). Bars A–C: 20 µm. (D) Immunoblotting analysis of laminin-332 purified from ATCC® CCL-15 cell media using the following reactants: sera from 2 control individuals (lanes 1, 2), patient serum (lane 3); a monoclonal antibody (K140) to the laminin β3 subunit (140 kDa) (lane 4); a mixture of antibodies to laminin α3 (BM165, 165 kDa) and γ2 (a homemade polyclonal antibody, 155 and 105 kDa) chains (lane 5). Patient serum reveals protein bands corresponding to the laminin α3 and β3 chains.
Fig. 3. Mutation identification, amino acid conservation and reverse transcriptase PCR (RT-PCR) analysis. (A) Sanger sequencing confirmation of compound heterozygous COL7A1 mutation c.410G>A in exon 3 (left panel, arrow, codon 137 underlined) and c.3674C>T in exon 27 (right panel, arrow, codon 1225 underlined). (B) Conservation of type VII collagen Arg137 residue (single letter code) between species (upper panel). Alignment of von Willebrand factor type A domains (lower panel) from paralogous proteins shows conservation between residues Arg137 and Arg1597 from human COL7A1 and vWF proteins, and among α1 chains from collagens XX, XII and XIV. (C) Electrophoretic gel of RT-PCR over the site of the c.3674C>T (left panel) and sequencing of the gel-purified cDNA products (right upper and lower panels) identify the expected full-length mRNA (305 bp) and an aberrant shorter transcript (244 bp) lacking 51 nucleotides (shaded area in the upper box). bp: base pair; C: control; P: patient.
Sequential treatments with prednisone (1 mg/kg/day) and colchicine (2 mg/day) induced only minimal clinical improvement. Thereafter, the patient refused any additional therapy.
We report here an Italian female patient with a missed “nails-only” subtype of RDEB since childhood, complicated by mechanobullous EBA in adulthood. Although the nails-only phenotype is usually a dominant trait, recessive transmission has also been reported (11). It has rarely been described in the literature, most likely because of negligible clinical implications and lack of overt skin fragility. Our findings confirm that, in sporadic cases with minimal disease symptoms, such as the “nails-only” subtype, DEB diagnosis can be overlooked. In our patient the suspicion of a mild DEB variant was only raised following the onset of skin and oral blistering due to the development of the autoimmune disease. Indeed, our results demonstrate for the first time that EBA can develop in sporadic patients with RDEB. Taken together with a previous report of EBA manifesting in a patient with DDEB (12), they point to the usefulness of performing DIF in patients with DEB showing unforeseen skin symptom worsening or modification. Conversely, personal history should be evaluated carefully in patients with EBA, as it may prompt mutation screening to unveil mild variants of RDEB.
The COL7A1 mutations identified in our patient, combined with autoantibody reactivity to C7, most likely contribute to the altered morphology and function of anchoring fibrils and, thus, to the development of skin fragility. The p.Arg137Gln replaces a strictly conserved residue within the vWFA1. The vWFA motif is a theme common to adhesive proteins and typically present in the vWF protein, the deficiency of which causes von Willebrand disease (vWD), an inherited bleeding disorder (13). Interestingly, missense mutations targeting vWF at the homologous residue Arg1597 (Fig. 3B), such as p.Arg1597Gln, cause vWD type 2a subtype (vWF variant registry: http://www.shef.ac.uk/vwf/). Thus, an arginine at this position seems important for vWFA domain folding and/or function. Regarding the c.3674C>T mutation, its main effect is the p.Ala1225_Gln1241 deletion consequent to partial exon skipping. This 17-amino acid segment bridges the vWFA2 to the cysteine-rich region and is adjacent to the cystine knot motif (CX3CP) used to form interchain disulphide bridges N-terminally to the collagenous domain. The function of the cystine knots is to prevent unfolding of the triple helix from the N-terminus (14). The p.Ala1225_Gln1241del may thus perturb either the vWFA2 architecture or the formation and/or topology of the cystine knots. These effects may impair binding to other collagens or favour partial unfolding of the N-terminal end of the triple helix (2, 3).
Circulating C7 autoantibodies are detectable in more than half of patients with RDEB and occur independently from COL7A1 mutation type, C7 abundance and patient age (6, 7, 12). However, direct and indirect immunofluorescence are usually negative, indicating that, in most cases, circulating autoantibodies are not pathogenic. The influence of genetic and/or environmental factors is thus probably crucial for development of EBA in DEB. It is also possible that particular COL7A1 variants in the NC1 and triple helix represent a risk for development of EBA in selected patients (12). Mutant, partially unfolded, NC1 subdomains can be thermolabile and more susceptible to protease cleavage, possibly resulting in the generation of neo-epitopes (15). In addition, compound heterozygous COL7A1 mutations, one of which has leaky effects on splicing, result in a mixture of C7 homotrimers, which can be composed of all 3 chains mutated in either of 2 positions or assembled from various combinations of the mutant and wild-type monomers, or even formed by all 3 wild-type chains. Thus, the NC1 domain in our patient presents multifaceted to the immune system and might evoke an autoimmune response in time. Circulating autoantibodies to laminin-332 chains and bullous pemphigoid antigens were also present in patient serum. Indeed, many targets of autoimmune diseases are part of multi-protein complexes, such as the spliceosome in lupus (16). We hypothesize that, following the induction of a chronic autoimmune response to C7, intermolecular epitope spreading events lead to development of autoantibodies to laminin-332 and BP180, which are target molecules physically linked in a complex to C7 in the BMZ (1, 3, 17). A similar intermolecular epitope spreading event has been observed in EBA (18).
In conclusion, these findings highlight the role of anti-C7 autoantibodies in inducing EBA in a woman with RDEB. In parallel, the pathogenicity of previously undescribed COL7A1 mutations in RDEB nails-only subtype is shown, thus increasing our knowledge of genotype-phenotype correlations in DEB. Sequence variants altering the structure of NC1 subdomains may represent a risk for development of EBA in predisposed patients. However, the molecular mechanism through which these sequence variants induce EBA remains to be established. Finally, regular DIF screening in patients with DEB who present with worsening of skin fragility may reveal additional examples of DEB and EBA overlap and be used to assess the frequency of this association, with important therapeutic implications.
The authors would like to thank the patient for granting permission to publish this information, Giovanna Zambruno for critical reading of the manuscript, Naomi De Luca for skilful technical assistance and Maurizio Inzillo for artwork.


 Click to show fullsize
Click to show fullsize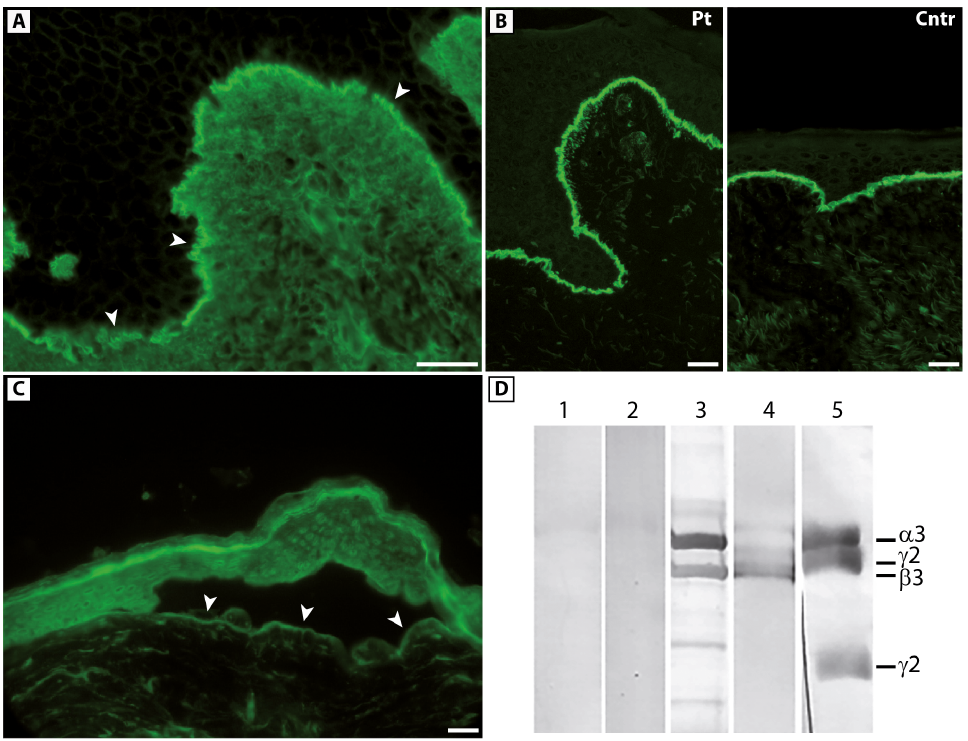 Click to show fullsize
Click to show fullsize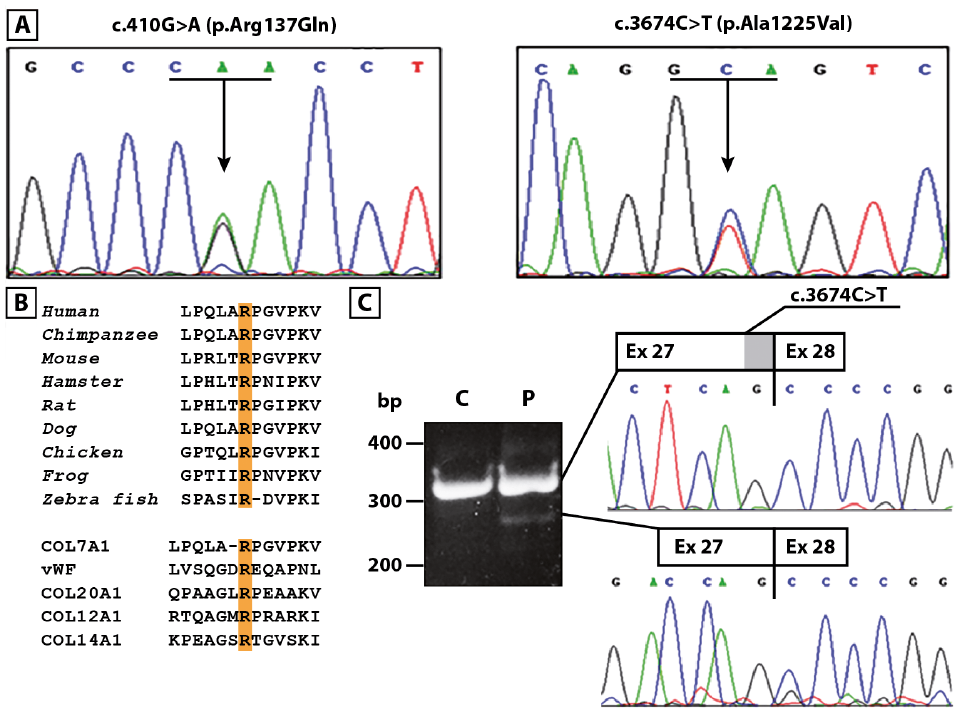 Click to show fullsize
Click to show fullsize