


Departments of 1Dermatology, and 2Internal Medicine, Teikyo University School of Medicine, 2-11-1 Kaga, Itabashi-ku, Tokyo 173-8606, Japan. E-mail: ytada-tky@umin.ac.jp
#These authors contributed equally to this work.
Accepted Jan 23, 2018; Epub ahead of print Jan 24, 2018
Adult-onset Still’s disease (AOSD) is a rare systemic inflammatory disease characterized by spiking fevers, increased white blood cell count, arthralgia and skin rash (1). The typical skin rash of AOSD is salmon pink-coloured, mildly itchy, and apparent during high fever. Histopathology of biopsy specimens from the eruption show dermal perivascular infiltrates of lymphocytes and neutrophils (2). Other than the typical rash, different types of skin manifestation of AOSD have been reported, such as dermatographism-like persistent eruption (2), violaceous to dusky red, scaly lichenoid papules (2), and intensely pruritic persistent erythema (3). These atypical eruptions are called persistent pruritic eruptions (PPE) (4). Here, we report 2 patients who were diagnosed as having AOSD with PPE showing peculiar histopathological findings. Prednisolone, 1 mg/kg/day, was not sufficient to resolve their systemic inflammation. The importance of PPE in diagnosing AOSD and predicting the disease outcome is discussed.
Case 1. A 75-year-old woman presented to the internal medicine department of our hospital with high fever (> 39°C), swollen painful knees, sore throat, and skin rash persisting for one month. Prior to presentation to the internal medicine department, treatment with oral aminobenzyl penicillin, 2,000 mg/day for 10 days, was not effective. Three weeks prior to admission, dark-red plaques with severe pruritus appeared on her chest, which persisted even when the fever subsided. Her body temperature was 37.4°C and she had cervical lymphadenopathy on admission. Dark-red to violaceous scaly plaques were seen on her chest (Fig. S1a). Laboratory findings revealed neutrophilia (WBC 6,600/μl, neutrophil 5,060/μl), anaemia (Hb 6.3 g/dl), thrombocytopaenia (10.0×104/μl), elevation of liver enzymes (AST 852 U/l, ALT 776 U/l, γGTP 397 U/l), hyperferritinaemia (167,998 ng/ml), and increased serum soluble interleukin-2 (IL-2) receptor (5,995 U/ml). Anti-nuclear antibody and rheumatoid factor were negative. Histopathology of biopsy specimens of the rash showed focal hyperkeratosis, flattening of rete ridges, and scattered dyskeratotic cells in the epidermis (Fig. S1b, c). Perivascular lymphocyte infiltrates and nuclear debris were observed around dermal blood vessels (Fig. S1d). The patient was diagnosed as having AOSD according to the diagnostic criteria (5). She was initially treated with prednisolone, 1 mg/kg/day for 10 days. However, haemophagocytosis, disseminated intravascular coagulation and interstitial pneumonia emerged during her clinical course. The patient was then treated with 7 sessions of plasmapheresis, together with cyclosporine, 2 mg/kg/day, and an interleukin-6 receptor antagonist tocilizumab (Chugai Pharmaceutical, Tokyo, Japan), 8 mg/kg/day, and finally her condition was controlled.
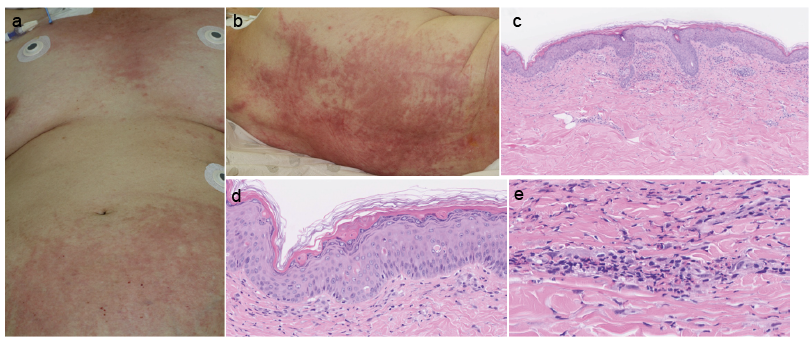
Case 2. 77-year-old woman presented to the emergency room of our hospital with a history of high fever (38–39°C) and intensely pruritic skin rash persisting for one week. She had received intravenous cefepime for 5 days at the nursing home, but this treatment failed to resolve her symptoms. On admission, her body temperature was 38.5°C and she did not have arthritis. Oedematous erythema with severe pruritus showing linear configurations was seen on her chest, abdomen, back, and inguinal region (Fig. 1a, b). The erythema persisted even at the times when she had normal body temperature. Laboratory findings revealed leukocytosis with neutrophilia (WBC 11,400/μl [normal 3,000–9,800], neutrophil 10,602/μl [40–70%]), anaemia (Hb 9.5 g/dl [10.2–15.5]), thrombocytopaenia (3.7×104/μl [15–38×104]), elevated liver enzymes (AST 73 U/l [1–32], ALT 26 U/l [6–35], γGTP 36 U/l [8–33]), hyperferritinaemia (43,865.7 ng/ml [6.4–167.1]) and increased serum soluble IL-2 receptor (3,616 U/ml [154–472]). Anti-nuclear antibody and rheumatoid factor were negative. The erythrocyte sedimentation rate (ESR) was increased (26 mm/h). Anti-nuclear antibody, anti-Jo-1 antibody and rheumatoid factor were negative. Histopathological examination of biopsy specimens of the skin rash showed flattening of rete ridges (Fig. 1c) and scattered dyskeratotic cells in the epidermis (Fig. 1d). Lymphocytes, neutrophils, histiocytes and nuclear debris were seen around superficial dermal blood vessels (Fig. 1e). The patient was diagnosed as having AOSD according to the diagnostic criteria (5). She was initially treated with prednisolone, 1 mg/kg/day for 10 days. However, haemophagocytosis, disseminated intravascular coagulation and liver enzyme elevation persisted. Additional cyclosporine was administered and 7 sessions of plasmapheresis were performed to reduce the dose of prednisolone to 0.3 mg/kg/day. Although AOSD was under control, the patient died 7 months after admission due to concomitant interstitial pneumonia.
Fig. 1. Clinical appearance and histopathology of Case 2. (a) Persistent oedematous erythema on the chest and the lower abdomen. (b) Persistent oedematous erythema with linear configurations on the back. (c) A skin biopsy specimen showing flattening of rete ridges and perivascular infiltrate in the dermis (HE ×40). (d) Dyskeratotic cells in the epidermis (HE ×200). (e) Perivascular infiltrate consisting of neutrophils, histiocytes, lymphocytes and nuclear debris (HE ×400).
Both patients fulfilled the criteria of AOSD (5) and had an extremely elevated serum ferritin level. Autoimmune diseases, malignancy, and infectious diseases were ruled out.
Two kinds of skin symptoms of AOSD have been reported: the typical salmon-pink rash and other atypical ones (6). The typical rash appears only when the body temperature increases, and usually appears without pruritus. On the other hand, atypical rash emerges during the early phase of AOSD, and its emergence does not depend on body temperature.
Atypical rash was first reported in a patient with AOSD by Lübbe et al. (7) in 1999. They described pruritic rashes, which consisted of oedematous erythema or various-coloured scaly papules, sometimes with a linear configuration. The rash could clinically resemble prurigo pigmentosa (8). Histopathology of the atypical rash was reported to have a characteristic pattern of dyskeratosis present mainly in the upper layers of the epidermis (2), a sparse superficial dermal infiltrate often containing neutrophils, but without vasculitis (2, 9) and neutrophilic epidermal and adnexal epitheliotropism (10). Furthermore, Lee et al. (4) described these rashes as persistent pruritic eruptions (PPE) and suggested that the diagnosis of PPE can be particularly helpful for reaching the diagnosis of AOSD when the clinical manifestations do not yet meet Yamaguchi’s criteria (5). Ohashi et al. (1) also reported that PPE could be an initial manifestation of AOSD. Therefore, PPE is pathologically distinct and may be a specific enough manifestation to contribute to the diagnosis of AOSD.
PubMed-MEDLINE was searched for English- and non-English-language articles published between 1980 and 2014 for case reports or population-based studies of persistent atypical skin rash in patients with AOSD. As shown in Table SI, 82 cases were found (4, 6, 7, 11). Sixty-three (85%) out of the 74 cases, including our patients, were female. The age range was 16–82 years old. The colour of skin rash varied from erythematous to brown, and was less commonly violaceous. Linear configurations were also mentioned in some cases. Histopathologically, many of those cases showed epidermal dyskeratotic keratinocytes and non-specific perivascular inflammatory infiltrates usually with lymphocytes and neutrophils. In most of the cases, skin rashes were resolved with the use of systemic prednisolone or intravenous pulsed methylprednisolone. However, some cases required more potent systemic therapies, such as hydroxychloroquine, cyclosporine, methotrexate, anakinra or tocilizumab. Both of our cases also had these common histopathological findings. These findings strongly suggest that our patients had PPE with AOSD. It has also been proposed that AOSD patients who present with PPE accompanied by severe systemic inflammation, are resistant to systemic corticosteroid therapy (1). Indeed, many AOSD cases with PPE required additional higher doses of prednisolone and/or other immunosuppressive agents (Table SI) (2, 12). Our 2 cases also required methylprednisolone pulse therapy, immuno-suppressive agents, biologic agents and plasmapheresis. We have to keep in mind that AOSD can be complicated and life-threatening due to haemophagocytosis, disseminated intravascular coagulation, or multi-organ failure, and PPE could be a sign of these complications.


 Click to show fullsize
Click to show fullsize