


1Department of Dermatology, Changwon Fatima Hospital, Changwon, 2Department of Life Science, Dongguk University-Seoul, Goyang-si and 3Department of Dermatology, College of Medicine, The Catholic University of Korea, Seoul, Korea
#These authors contributed equally to this work.
This study analysed genomic mutations in basal cell carcinoma using whole exome sequencing of DNA specimens obtained from 20 Korean patients. Histological evaluation determined that 15 (75%) were low-risk basal cell carcinomas, and 5 (25%) were high-risk basal cell carcinomas. Seventy-five percent of the basal cell carcinomas harboured somatic mutations in hedgehog pathway genes (PTCH1, 40% and SMO, 50%) and 45% harboured mutations in TP53. LRP1B was the most frequently mutated gene in high-risk basal cell carcinomas, SMO was the most frequently mutated gene in low-risk basal cell carcinomas. Specifically, LRP1B, ROS1, PTCH1, KMT2C, NSD1 and ARID1A mutations were more frequent in high-risk basal cell carcinomas than in low-risk basal cell carcinomas. However, copy number gains of the ROS1 gene were observed only in low-risk basal cell carcinomas. Other basal cell carcinoma related genes found in this study include: KDR, KMT2D, FAT1, FAT4, GRIN2A, ERBB4, NOTCH2, PDE4DIP, TET1, ZFHX3 and PREX2. These results provide insight into basal cell carcinoma in non-Caucasians.
Key words: basal cell carcinoma; whole exome sequencing; skin cancer; genetics; histopathology; mutation.
Accepted Apr 29, 2021; Epub ahead of print Apr 30, 2021
Acta Derm Venereol 2021; 101: adv00458.
doi: 10.2340/00015555-3820
Corr: Young Bok Lee and Dong Soo Yu, Department of Dermatology, Uijeongbu St. Mary’s Hospital, 271 Chunbo Street, Uijeongbu, 07345, Korea. E-mails: lyb80@catholic.ac.kr; frankyu123@hotmail.com
This study analysed genomic mutations of basal cell carcinoma using whole exome sequencing of DNA from 20 Korean patients. Histopathological evaluation found that 15 (75%) were low-risk basal cell carcinomas, and 5 (25%) were high-risk basal cell carcinomas. Seventy-five percent of basal cell carcinomas harboured somatic mutations in hedgehog pathway genes (PTCH1, 40% and SMO, 50%) and 45% harboured mutations in TP53. LRP1B was the most frequently mutated gene in high-risk basal cell carcinomas and SMO was the most frequently mutated gene in low-risk basal cell carcinomas. These results provide insight into basal cell carcinoma in non-Caucasians.
Basal cell carcinoma (BCC) is the most common human cancer worldwide, accounting for up to 80% of all non-melanoma skin cancers (1). The incidence of cutaneous BCC is 2.75 million cases worldwide (2) and is increasing by up to 10% each year (3). Although BCC is commonly diagnosed in Caucasians, it is known to have low incidence rate in Asian-ancestry populations, including Korea. The incidence rate of BCC in Korea during 1999–2014 was 2.45 per 100,000 in men, and 2.07 per 100,000 in women, respectively (4). Incidences of BCC per 100,000 population in other races have been reported as follows: black men (1), black women (2), Chinese men (6.4), Chinese women (5.8), Japanese (15–16.5), Japanese residents of Kauai, Hawaii (29.7), New Mexican Hispanic women (113), New Mexican Hispanic men (171), Caucasian men (250), Caucasian women (212), and Caucasians in Kauai, Hawaii (185–340) (5). BCC is seldom fatal, and has a very low metastasis rate (0.003–0.55%) (6), but it results in extensive morbidity through local tissue destruction if left untreated. Considering the frequent occurrence of BCC on the face, it imposes a significant therapeutic challenge and represents a growing public health problem.
Recent research has provided insight into the pathogenesis of BCC, which is thought to involve both environmental and genetic factors. Exposure to ultraviolet (UV) radiation is the most common risk factor for BCC in all racial groups. Other possible risk factors for BCC include exposure to ionizing radiation, or arsenic; immune suppression; male sex; and age. The major host susceptibility risk factors include lighter pigmentation characteristics, such as fair skin, blond hair, and light eye colour (7). Several genetic mutations are associated with BCC development, such as those in TP53 and PTCH1. Loss-of-function mutation of tumour suppressor PTCH1 activates the sonic hedgehog signalling pathway, leading to BCC development (8). Tumour suppressor TP53, which is involved in cell cycle arrest and activation of programmed cell death, is also frequently inactivated in BCC and plays a role in disease development (8).
Most previous studies that have investigated genetic susceptibility to BCC have focused primarily on common single-nucleotide polymorphisms excluding rare variants; however, the findings are not sufficient to completely elucidate the genetics behind this complex disease. To examine the entire set of protein-coding genes, an increasing number of cancer studies are using whole exome sequencing (WES). Recently, Jayaraman et al. used WES on 10 Caucasian patients with BCC, and found that CSMD1, CSMD2, NOTCH1, NOTCH2, GRIN2A, and PREX2 were frequently mutated in BCC (9). Although many genetic loci associated with BCC have been identified by WES, most studies have been conducted on Caucasian populations, and little is known about genetic alterations in non-Caucasians, who have a low prevalence of BCCs.
This study performed WES in Korean patients in order to identify the mutational frequency, spectrum and type of these genes and to determine whether differences observed between different racial populations may be related to these genetic alterations. In addition, the study aimed to find the differences in genetic mutations between low-risk and high-risk BCCs. The findings may increase our understanding of the molecular events involved in the development and expression of BCC in non-Caucasian patients.
Clinical samples
BCC samples and peripheral blood were obtained from 20 non-Caucasian patients of Korean ancestry living in Korea who were diagnosed with BCC of the face and had undergone excisional surgery at the Dermatosurgery Clinic at Uijeongbu St Mary’s Hospital. Lam et al. suggested that BCCs in Chinese persons might be due to arsenic (10), but that does not seem to be the case in the current study. Arsenic-related BCCs are often multiple, and most frequently found on the trunk (11), but our sample set comprised 20 sporadic cases on the face. The collection did not comprise tumours from patients with nevoid basal cell carcinoma syndrome (Gorlin syndrome) or xeroderma pigmentosum, and none of the patients were immunosuppressed or in post chemotherapy.
Twenty BCC tissues from 20 different patients were included. All tissues and blood samples were stored in Uijeongbu St Mary’s Hospital Human Biobank under cryogenic conditions. The biospecimens for this study were provided by The Catholic University of Korea, Uijeongbu St Mary’s Hospital Human Biobank. Anonymized and de-identified information was used for analyses. The study was approved by the Institutional Review Board at Uijeongbu St Mary’s Hospital, The Catholic University of Korea (UC17TNSI0078). The principles of the Declaration of Helsinki were followed, and patients provided written informed consent before enrollment and sample collection. Only tumours with > 90% tumour area on histological sections were chosen for WES.
Histopathological analysis
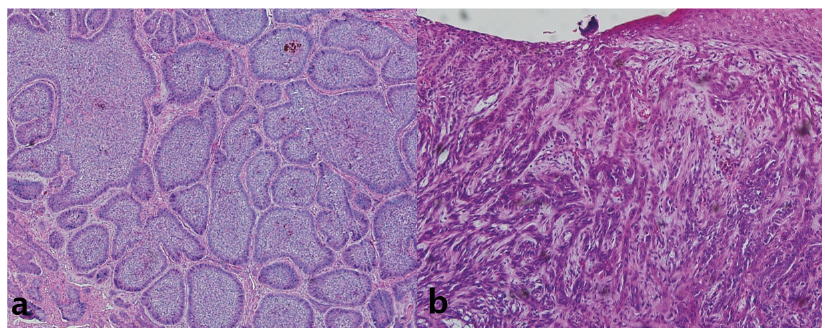
Histopathological analysis was performed on the BCC samples. Histological subtypes of BCC stratified by the risk of recurrence described in the current World Health Organization (WHO) classification are as follows: low-risk BCCs (nodular, superficial, pigmented, fibroepithelial, infundibulocystic), high-risk BCCs (infiltrating, basosquamous, sclerosing/morphoeic, sarcomatoid, micronodular) (12, 13). According to WHO classification, 15 (75%) were low-risk BCCs, and 5 (25%) were high-risk BCCs. A recent European BCC study also confirmed the predominance of the low-risk type BCCs (71%), and the incidence of high-risk BCCs was 29%, which was similar to the results of the current study (14). The representative histological findings of each subtype are shown in Fig. 1. Haematoxylin and eosin (H&E) stained histology for all samples are shown in Fig. S1.
Fig. 1. Representative histology of basal cell carcinoma (BCC) samples. (a) Low-risk BCC, (b) high-risk BCC. Magnification, 100×.
Whole exome sequencing and bioinformatic analyses
DNA from 20 BCC tissues was separately analysed by WES using the Agilent SureSelect Human All Exome 50Mb Kit (Agilent Technologies, Santa Clara, CA, USA). All samples were matched with blood samples from the respective patient to identify somatic mutations. Using the Illumina HiSeq2000 platform to generate 101-bp paired-end reads, the Burrows-Wheeler aligner, version 0.7.15 (15) was used to align the sequencing reads onto the human reference genome (hg19). The aligned sequencing reads were evaluated using Qualimap (16), and PCR duplications were marked by Picard-tools-2.7.1 (http://picard.sourceforge.net/), followed by data cleanup with GATK version 3.6 (17). BCC samples were sequenced with a mean coverage of 95X. Somatic variants were then identified by MuTect (18). The ANNOVAR package (19) was used to select somatic variants located in the exonic sequences and predict their functional consequences. High-confidence mutations were selected with a threshold read depth ≥10 and a variant allele frequency ≥ 5%. Copy number alterations (CNA) were identified by EXCAVATOR (20). The mutations identified in this study were compared with the somatic mutations in BCCs reported in the COSMIC database (https://cancer.sanger.ac.uk/census), which is a catalogue of somatic mutations in cancer. Then, among the genes identified in the current study, which are also frequently mutated in the COSMIC database, the top 19 genes were chosen according to the mutation frequency and an oncoprint was constructed for them. Mutational signatures of each sample were extracted by R package “deconstructSigs” (21), based on signatures previously identified by the Wellcome Sanger Institute (https://cancer.sanger.ac.uk/cosmic/signatures).
Mutational burden analysis
WES was performed on 20 BCCs and tumour mutational burden (TMB) was calculated, which is defined by total non-silent somatic mutation counts in coding regions. A total of 19,805 mutations were uncovered in the “protein-coding” DNA regions across all samples. Among the overall mutations,12,919 were non-silent (non-synonymous SNV, gain or loss of stop codon, frameshift or in-frame indel, or change in splice site). More than half of the non-silent mutations were non-synonymous SNV (Fig. 2a). The mean TMB of the Korean patients in the current study was 12.9 mutations/megabase (Mb) (median 9.7), ranging from 1.1 to 38.5/Mb. No significant difference in TMB status was observed between low-risk BCCs and high-risk BCCs in the current cohort. The mean TMB was 12.6 (median 9.6) for low-risk BCCs and 13.6 (median 9.9) for high-risk BCCs, respectively.
Fig. 2. Somatic SNV profiles. (a) Total numbers of non-synonymous SNVs (red), synonymous SNVs (blue), stop-gain SNVs (yellow), and stop-loss SNVs (green) for each of the 20 basal cell carcinomas (BCC) in this study. (b) Mutation types in the 20 BCC samples.
Basal cell carcinoma driver mutation analysis
The mutations identified in this study were compared with known BCC somatic mutations reported in the COSMIC database. Then, among the genes that are also detected in the COSMIC database, the top 19 genes were chosen according to the mutation frequency, and an oncoprint was made for them showing their genetic alterations (Fig. 3). Details of the types and frequencies of genetic alterations of each gene are shown in Table I. In the current study, 75% of BCCs harboured somatic mutations in hedgehog pathway genes (PTCH1, 40% and SMO, 50%) and 45% harboured mutations in TP53. Twenty BCCs in the current study were sporadic BCCs and no cases harbouring two hits in PTCH1 or TP53 were observed. Several known cancer-related genes PREX2 (10%), GRIN2A (25%), NOTCH2 (25%), ARID1A (25%) were recurrently mutated in BCCs with low frequency in the current study. In addition, many other cancer-related gene mutations (LRP1B (50%), ROS1 (40%), KDR (45%), KMT2D (40%), FAT1 (30%), FAT4 (30%), NSD1 (30%), KMT2C (30%), ERBB4 (15%), PDE4DIP (15%), TET1 (10%), ZFHX3 (10%)) were identified in the current study. The hotspot mutation-containing genes (n = 96) found in more than 2 BCCs including MYCN and CSMD3 are listed in Table SI.
Fig. 3. Oncoprint of somatic alterations for the top 19 basal cell carcinoma (BCC) genes. CN: copy number.
Table I. Types and frequencies of genetic alterations of top 19 basal cell carcinoma (BCC) genes
In high-risk BCCs, the profile of driver mutations was different from that of low-risk BCCs. Specifically, in high-risk BCCs, LRP1B, ROS1, PTCH1, KMT2C, NSD1, ARID1A mutations were 1.3, 1.8, 1.8, 1.5, 1.5 and 2 times more frequent than in low-risk BCCs, respectively. In low-risk BCCs, SMO mutation was 1.3 times more frequent than in high-risk BCCs. However, the result did not reach statistical significance, due to the small sample size. Also, non-synonymous mutations in NOTCH2, KMT2D, GRIN2A, FAT1, FAT4 and ERBB4 were not found in high-risk BCCs, but only in low-risk BCCs.
Copy number alteration analysis
A total of 178 CNAs were identified from the 20 BCC samples, with a median of 4 per case, including 113 copy number (CN) gains and 65 CN losses (Fig. S2). Whereas SMO showed aberrant activation with CN gain, 75% of the PTCH1 mutations and 44% of the TP53 mutations were stop-gain mutations, consistent with their roles as tumour-suppressor genes.
Whereas there were no stopgain or non-synonymous mutations of GRIN2A in high-risk BCCs, those mutations were seen in 33% of low-risk BCCs. CN gains of ROS1 gene were observed only in low-risk BCCs (27%). CN gain of NSD1 gene was observed in one low-risk BCC (5%). CN gain of both SMO and KMT2C were observed in one low-risk BCC, and one high-risk BCC. Interestingly, CN loss of genes was shown only in high-risk BCCs. CN loss of PTCH1, KDR, FAT1, FAT4 and NOTCH2 genes was detected in 20% of high-risk BCCs (Fig. 3).
Mutational signature analysis
As shown in Table II, UV-associated mutational signatures 7a and 7b were the most dominant signatures, regardless of histological classification. Signature 31 was seen in only 1 BCC patient. The proposed aetiology of signature 31 is prior chemotherapy treatment with platinum drugs, but that was not the case in this patient, who had never previously received chemotherapy. C/G>T/A transitions, a characteristic “UV signature” mutation, were the most common mutations in our BCCs, confirming that UV radiation is the primary cause of mutations in BCC (Fig. 2b). Both C/G>A/T and C/G>G/C transversions (tobacco signature, oxidative damage-associated mutation signature) were found in low proportion in the BCCs in the current study (Fig. 2b) (22, 23).
Table II. Mutational signature analysis
This study used WES of sporadic BCCs from 20 Korean patients to investigate differences in genetic mutations between low-risk and high-risk BCCs. Whereas LRP1B, ROS1, and PTCH1 were the most frequently mutated genes in high-risk BCCs, SMO was the most frequently mutated gene in low-risk BCCs. Amplification of chromosome 6 was identified only in low-risk BCCs that contained ROS1 proto-oncogene at 6q22. Gains associated with chromosome 6 have been of interest in previous studies, especially since this chromosome contains a number of genes associated with cancer, such as tumour suppressor gene CDKN1A (24) and oncogenes including VEGF and AP2 (25). Although the differences in profile of driver mutations between low-risk and high-risk BCCs did not reach statistical significance, due to small sample size, exploratory analysis of the results suggests a link between oncogenic mutations in the genes and histological subtype, and suggests that tumorigenic pathways might vary among the BCC subtypes.
The results were compared with studies of Caucasian populations. The mean TMB, which is a measurement of the number of non-synonymous mutations carried by tumour cells, of BCCs in the Korean patients was low (12.9/Mb) in comparison with BCCs in Caucasians (65–76 mutations/Mb) (9, 26). Also, the comparison of gene mutation profiles between Caucasians and Koreans showed racial differences. While the mutation frequency of SMO (50%) appears higher than that of PTCH1 (40%) in the Korean patients, SMO mutations occurred in approximately 10%–20% of BCCs, and PTCH1 mutations occurred in approximately 70% of BCCs in previous studies from Caucasian population (26, 27). This suggests a possible non-random nature of these mutations, or that the mutation profile varies in different patient populations. Therefore, we speculate that SMO might be a more common gene driver in BCC development in the Korean population. TP53 showed similar mutation frequencies between Caucasians (40%, 61%) and Koreans (45%) (26, 27). ARID1A and NOTCH2, being altered in a loss of function manner, are presumed to be tumour suppressor genes in BCC (28, 29), and their mutation frequencies did not show significant differences between the Korean patients in the current study (25%, 25%) and Caucasians (26%, 26%) (26). GRIN2A and PREX2 mutation frequencies appear to be lower in BCCs from Korean patients (25%, 10%) compared with Caucasians (50%, 58%) (9). GRIN2A, which encodes a member of the glutamate-gated ion channel protein family, is a tumour suppressor in melanoma, but the involvement of glutamate signalling in BCCs is still unclear (30). PREX2, which encodes a regulator of PTEN and the PI3K-Akt pathway, is mutated in various other cancers, with mostly gain of function mutations that lead to promoted cell proliferation (31–33). Although activation of the PI3K/AKT signalling pathway has also been reported in BCC, the role of PREX2 in BCC has not yet been elucidated. LRP1B was the most frequently mutated gene found in 50% of BCCs in the Korean patients in the current study, although it has not been reported previously in Caucasians. Expression of LRP1B, a tumour suppressor gene, inhibits cancer cell growth by increasing Stat3 phosphorylation and p21C1P1 level, as well as by inactivating the phosphorylation of SRC (34), and inactivation of LRP1B is known to be involved in development of various cancers, including melanoma (35–38). It seems to be a candidate BCC driver in Korean patients. Among hotspot mutation-containing genes, MYCN mutation rate (15%) was lower than that in a Caucasian study (30%) (26). BCC susceptibility locus at 2q24 in the vicinity of the MYCN region supports the implication of MYCN in BCC (26). Aberrant MYC regulation can lead to increased cell proliferation and is commonly observed in cancers, including neuroblastoma, medulloblastoma, prostate cancer and lung cancer (39). Also, CSMD3 mutation rate (10%) in the Korean patients in the current study was significantly lower than that of Caucasians (58%) (9). CSMD3 is a homolog with great structural similarity of complement inhibitor CSMD1, which is a putative suppressor gene of SCC and BCC.
In conclusion, this study describes a complex network of genes involved in development of BCC in non-Caucasian Korean patients using WES. The genetic landscape of BCC is highly complex, but, in the therapeutic strategy of BCC, surgery (either curative or palliative) remains the primary option. Analyses comparing the WES results from the current study with WES data from Caucasian patients have found discordance of somatic mutations in genes between Caucasians and non-Caucasian Korean patients with BCC. These observations may be related to the different BCC tumorigenesis between different racial populations. The most important limitation of this study was its small sample size. The results need to be confirmed in more BCC samples, especially with normal tissue control. Therefore, multi-centre studies with a larger number of samples are required. In addition, it remains important for future studies to include diverse patient populations, including both Caucasians and non-Caucasians. Despite the study limitations, these findings provide insight into the differences between non-Caucasian and Caucasian patients with BCC, and have practical implications for further research.
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2019R1F1A1056601).
The authors have no conflicts of interest to declare.


 Click to show fullsize
Click to show fullsize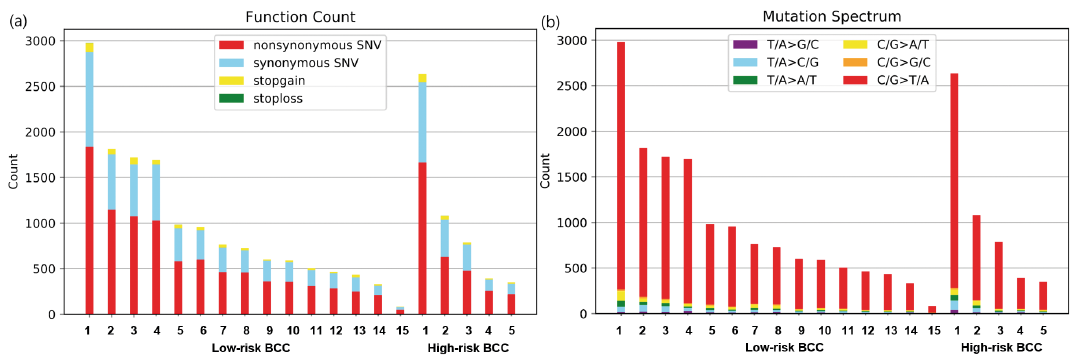 Click to show fullsize
Click to show fullsize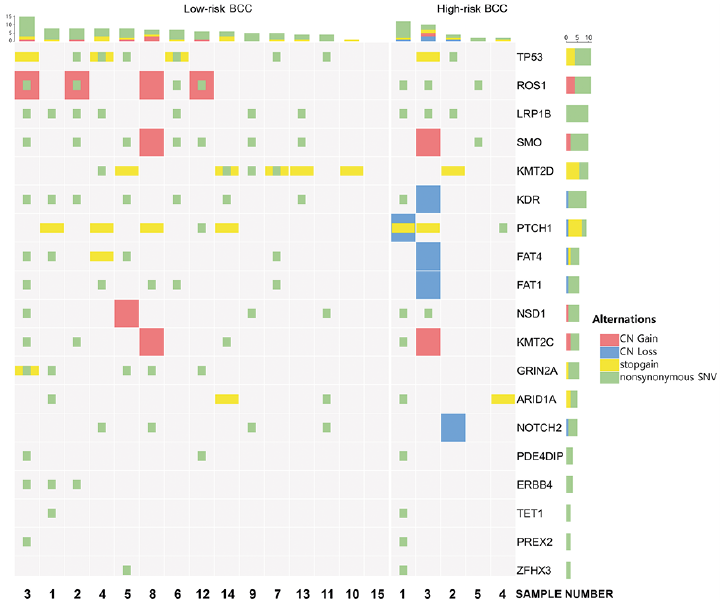 Click to show fullsize
Click to show fullsize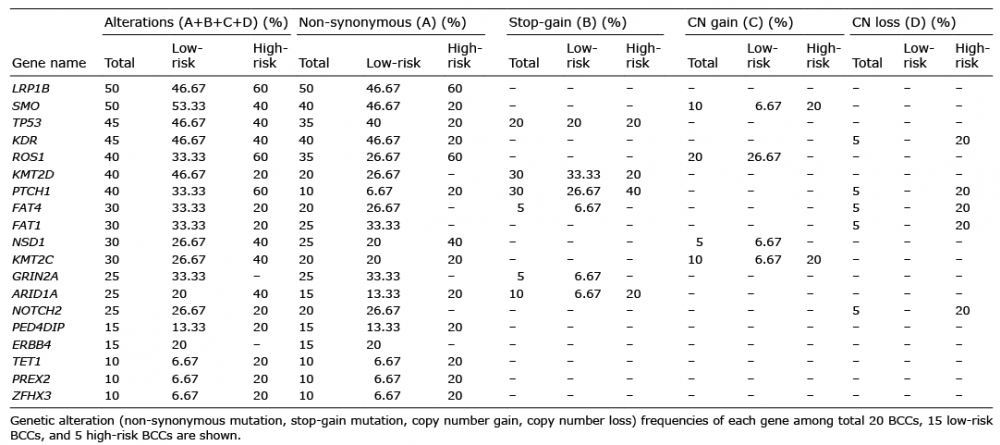 Click to show fullsize
Click to show fullsize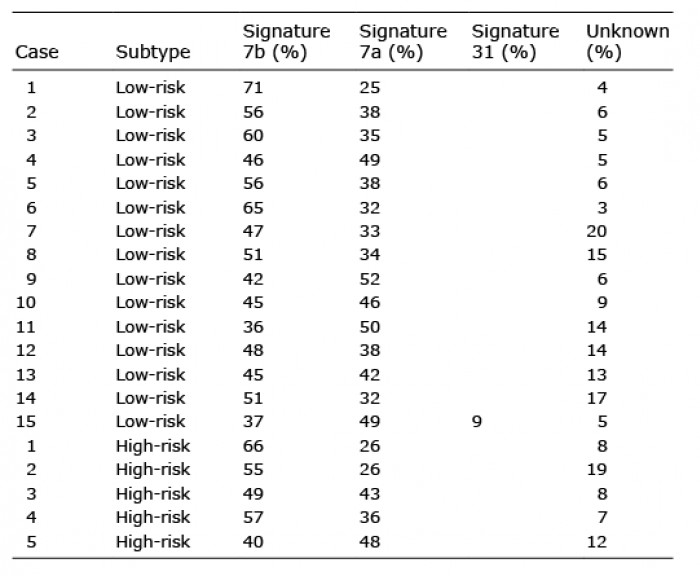 Click to show fullsize
Click to show fullsize